Clonal evolution monitoring in cancer with liquid biopsy

In the last part of our blog series, we will focus on the process by which subclones in tumors change over time and how liquid biopsy can be applied in monitoring clonal evolution and heterogeneity in tumors.
Cancer is a dynamically changing disease. During the course of the disease tumors become more heterogeneous due to clonal evolution, genetic diversification, and clonal selection. As a result of this heterogeneity, the tumor mass might include diverse groups of cells, harboring unique molecular signatures with distinct sensitivity to treatment (Dagogo-Jack and Shaw, 2017).
Cancer development can be considered an evolutionary process, which in many respects mirrors Darwin’s theory about species evolution. Species evolve by mutation and selection, acting on individuals in a population; tumors evolve by mutation and selection, acting on individual cells in a tissue. Cancer progression occurs when a cell clone acquires a number of genetic changes over time, escapes developmental constraints that would allow the maintenance and renewal of tissue structures and would help suppress harmful cell expansion. (Casás-Selves and DeGregori, 2011).
During cancer progression and treatment, multiple subclonal populations of tumor cells compete with one another, with selective pressures leading to the emergence of predominant subclones that replicate and spread most proficiently and are least susceptible to treatment. This evolutionary process in cancer is driven by two major forces ( Casás-Selves and DeGregori, 2011, Siravegna et al., 2017):
1. Mutation
Genetic variation in somatic cells promotes the acquisition of mutations in oncogenes and tumor suppressor genes leading to cancer development.
2. Selection
Factors associated with cancer development, such as aging and exposure to damaging carcinogens reduces general cellular fitness in healthy cells, therefore allowing opportunity to the selection for cells that gained adaptive oncogenic mutations with increased cellular fitness relative to competing cells, leading also to cancer progression.
While there are therapies that can eradicate already developed cancer cell populations, unfortunately, these treatments can also create powerful selective pressure that will allow the emergence of new clones that acquired resistance to the treatment regimen (Fig. 1.), leading to the re-emergence of treatment-refractory disease (Casás-Selves and DeGregori, 2011).
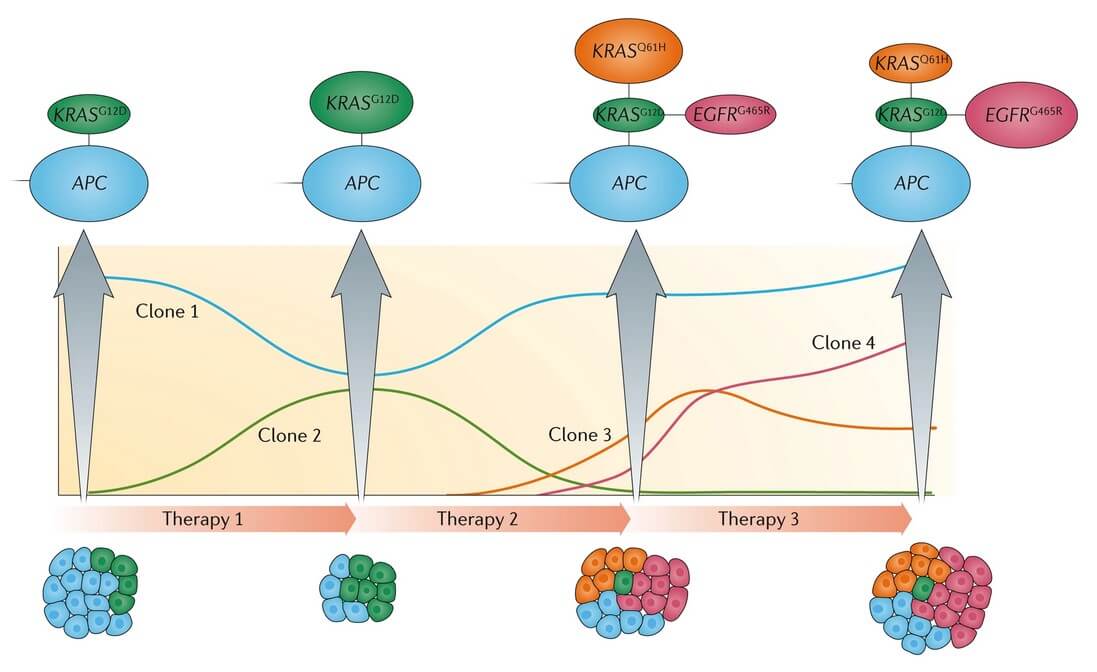
Fig.1. Monitoring clonal evolution using liquid biopsy. Schematic view of the dynamic gene5tic changes in subclones harboring different mutations in a patient with metastatic colorectal cancer, treated with targeted therapy (anti- Epidermal Growth Factor Receptor (EGFR) antibodies) (Siravegna et al., 2017).
Many studies indicate that clonal evolution in cancer is complex and leads to high heterogeneity (Bianchi et al., 2014). For instance, studies focusing on the genetic and molecular mechanism underlying clonal evolution suggest that myeloid malignancies including acute myeloid leukemia (AML), myelodysplastic syndrome (MDS), multiple myeloma (MM), myelodysplastic syndrome (MDS), and chronic lymphocytic leukemia (CLL) show branched pattern of clonal evolution (Ferrando and López-Otín, 2017), in which multiple distinct subclones co-exist. Subclones that acquired an additional mutation associated with treatment resistance (TP53) or disease progression (NRAS, KRAS) may be detected months before clinical changes become apparent (Silva-Coelho et al., 2017). Multiple studies have demonstrated also the link between subclonal diversification and adverse clinical outcome. Higher clonal diversity is associated with greater disease severity and poorer overall survivor (Turajlic et al., 2019).
Solid tumors, including lung, ovarian, liver, prostate cancer, etc. are also extensively studied to reveal acquired genomic alterations in subclonal tumor cells, responsible for the failures of targeted therapies and for the arise of drug resistance (Roper et al., 2020, Shlush and Hershkovitz, 2015, Testa et al., 2018, Zhang et al., 2022).
Genetic events and molecular mechanisms underlying cancer evolution have been difficult to explore, but now with liquid biopsy-based technologies researchers are able to reconstruct steps in tumor progression and identify main mutational drivers. Non-invasive, real-time, and longitudinal analysis of tumor-derived genetic materials are promising strategies for addressing the shortcomings of tissue sampling and have considerable potential to dissect the mutational complexity of cancers, contributing to drug resistance. Analysis of the genetic material obtained from circulating tumor cells (CTCs), circulating exosomes, and circulating cell-free tumor DNA (ctDNA) shed by tumors has yielded promising results across several types of solid tumors (Dagogo-Jack and Shaw, 2017).
Liquid biopsy is a sensitive and highly informative method used in the identification of clinically relevant genomic alterations, in concordance with the findings of tissue biopsy sample analysis. CtDNA particularly has the advantage of detecting changes affecting therapeutic resistance earlier than it is visible on imaging. Because ctDNA originates from multiple metastatic sites, it also allows the identification of alterations that were not detected by tissue genotyping (Fig.2.) (Siravegna et al., 2017, Dagogo-Jack and Shaw, 2017).
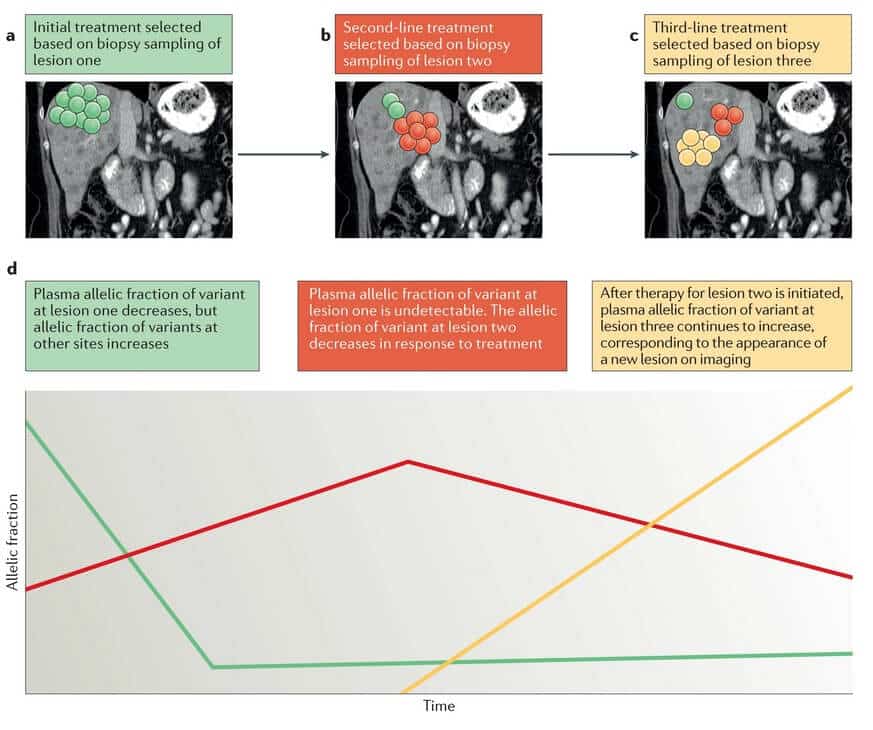
Fig.2. Longitudinal monitoring of genomic alterations in ctDNA has the potential to predict relapses before the emergence of disease relapse on imaging (Dagogo-Jack and Shaw, 2017).
A great illustration of why patients should be evaluated repeatedly during therapy with liquid biopsy is presented by Siravegna et al. (2015). The research group was able to identify the genetic mechanisms of primary resistance to anti-EGFR treatment in colorectal cancer patients. In the study ctDNA has been analyzed to genotype colorectal cancer and track clonal evolution at multiple points before, during, and after treatment with anti-EGFR antibodies. The abundance of KRAS-mutant clones that often emerge during EGFR blockade declined upon withdrawal of anti-EGFR antibody therapy, while the tumor regained drug sensitivity. Patients who would benefit from rechallenged treatment with anti-EGFR antibodies showed pulsatile levels of mutant KRAS based on their ctDNA profiles.
Beyond colorectal cancer, evidence of successful rechallenge strategies with targeted therapies were investigated in patients with other tumor types, particularly melanoma or non-small cell lung cancer (Seghers et al., 2012, Hata et al., 2013, Hata et al., 2013), but the molecular evolution has not been documented using liquid biopsy approaches. (Siravegna et al., 2017) Clinical implementation will be possible if standardized procedures are defined and large validation studies are performed.
Research studies over recent decades have established that cancer development is inherently tied to our evolutionary history, in a complex manner. In cancer, mutations and selective pressure – key principles of clonal evolution – promote resistance to therapies. However, these same evolutionary principles can be applied to successfully manage cancer. Tumors with a mixture of multiple genotypically and phenotypically distinct cell populations will likely require targeting multiple resistance mechanisms by combination therapies. Thus, understanding clonal evolution with advanced longitudinal monitoring is essential and might facilitate the development of more-effective personalized therapies for cancer patients.
Recent blogs
A New Era in Liver Cancer Detection: The Promise of HepaAiQ
In this blog entry, we will explore the recent history, intriguing findings, and tools related to cfDNA fragmentomics.

Cell-free DNA Fragmentomics: A Promising Predictor of Cancer
In this blog entry, we will explore the recent history, intriguing findings, and tools related to cfDNA fragmentomics.
New developments in the field of circulating tumor cells (2024)
The blog post focuses on how researchers can produce more meaningful, applicable results that directly benefit human health.